低温肉制品中3 种食源性致病菌多重聚合酶链式反应快速检测方法的建立
李新福
1,2,王周平
1,李 聪
1,2,方伶俐
1,赵 颖
2,徐宝才
1,2,3,*
(1.江南大学食品学院,江苏 无锡 214122;2.江苏雨润肉食品有限公司,肉品加工与质量控制国家重点实验室,江苏 南京 211806;3.江苏省肉类生产与加工质量安全控制协同创新中心,江苏 南京 210095)
摘 要:针对低温肉制品中较易污染的沙门氏菌(Salmonella spp.)、单增李斯特菌(Listeria monocytogenes)和金黄色葡萄球菌(Staphylococcus aureus)等有害微生物,利用多重聚合酶链式反应(multiplex polymerase chain reaction,m-PCR)的方法,对3种菌同时快速检测,从而达到节省时间,提高效率的目的。使用沙门氏菌的invA基因、金黄色葡萄球菌的PrfA基因和单增李斯特菌nuc基因设计3对特异性引物,在确定特异性引物的基础上,将3种菌进行培养增菌后接种于低温肉制品中,过夜富集后收集并进行灵敏度检测,优化多重PCR反应体系并应用于实际样品的检测。结果表明:多重PCR方法在同时检测这3 种有害微生物时,m-PCR最佳反应条件如下:25 μL反应体系、10×PCR缓冲液2.5 μL、2.5 mmol/L dNTPs 2.0 μL、2.5 U/μL的DNA聚合酶0.5 μL及上、下游引物各0.5 μL、单增李斯特菌和金黄色葡萄球菌的模板1.0 μL、沙门氏菌的模板2.0 μL,加ddH
2O补足至25 μL,最佳退火温度为64 ℃。多重PCR快速检测方法适用于低温肉制品中沙门氏菌等3种食源性致病菌的检测,具有快速、灵敏、特异、简便等特点。
关键词:低温肉制品;多重PCR;快速检测;食源性致病菌
食源性致病是食品安全性问题重要的关注点,也是食品安全研究的一个重要方向。据统计,世界因食品污染而致病的人数已达数亿人,其中由生物性污染造成的人数占首位
[1],问题较为严重。对2001—2010年中国食源性疾病暴发监测与报告系统中资料显示,总计暴发5 021起,发病数为140 101人,致死数为1 427人,死亡率1.019%。查明致病因素的事件共4 243 起,占总数的84.51%;微生物性暴发事件数和发病人数最多,分别占总数的40.93%和56.39%
[2]。我国是肉制品生产和消费大国,由于肉制品中水分含量高、营养物质丰富,易受各种微生物的污染,滋生多种腐败菌和致病菌
[3]。目前,对食源性致病菌的检测多采用传统培养方法,因其步骤冗繁,耗时费力
[4],已不适应当前公共事件对检测方法实效性、准确性的发展需要。
生物学检测技术特别是聚合酶链式反应(polymerase chain reaction,PCR)技术作为食品微生物学的检测方法,近几年得到了迅速的发展
[5]。在普通PCR基础上发展起来的多重PCR(multiplex polymerase chain reaction,m-PCR)技术,是指在同一PCR反应体系中加入2 对或2 对以上的特异性引物,一次可扩增2 个或2 个以上的核苷酸片段,快速检测或鉴定2 种或2 种以上微生物的技术
[6-7],弥补了传统培养方法的不足,提高了检测效率及准确性,快速、经济、简便,是食源性致病菌检测技术的主要发展趋势
[8]。目前,m-PCR方法的建立主要集中于沙门氏菌(Salmonella spp.)、金黄色葡萄球菌(Staphylococcus aureus)、志贺菌(Shigella spp.)、大肠杆菌(E. coli)、单增李斯特菌(L. monocytogenes)等几种致病菌
[9-13],但在低温肉制品中的沙门氏菌、单增李斯特菌和金黄色葡萄球菌的m-PCR检测的相关研究和报道还较少。
本研究旨在建立一种低温肉制品中快速检测沙门氏菌等3种食源性致病菌的多重PCR方法,以期推广应用并有效减少致病菌的潜在危害。
1 材料与方法
1.1 材料与试剂
实验菌株:CICC21482肠炎沙门氏菌(Salmonella enteritidis) 中国工业微生物菌种保藏管理中心;ATCC6538金黄色葡萄球菌(Staphylococcus aureus)美国模式培养物研究所;单增李斯特菌(Listeria monocytogenes) 上海慧耘生物科技有限公司。实验菌株均用甘油管-80 ℃保藏。
培养基与试剂:营养肉汤培养基(NB培养基)、脑心浸液培养基(BHI培养基)、胰酪胨大豆酵母浸膏琼脂培养基(TSA-YE培养基) 北京陆桥技术股份有限公司;实验所用引物序列的合成和测序均由金斯瑞生物科技有限公司完成;离心柱型细菌基因组DNA提取试剂盒(DP320)、TaqDNA聚合酶、DNA Marker 北京天根生化科技有限公司;dNTP混合物、10×PCR缓冲液、6×上样缓冲液、50×TAE(Tris base- acetic acid-EDTA)缓冲液、琼脂糖 上海碧云天生物技术有限公司;GelRed染色剂 美国Biotium公司。
1.2 仪器与设备
Veriti 96-well型PCR仪 美国ABI公司;GelDoc-IT 310型凝胶成像分析系统 美国UVP公司;3K15型台式高速冷冻离心机 德国Sigma公司;BSC-1500IIA2-X型生物安全柜 山东博科生物产业有限公司;Milli-QDirect8型超纯水系统 美国Millipore公司;Revoc 379L型超低温冰箱 美国Thermo公司;Microone PMC-880型小型离心机 日本Tomy公司;LAB Dancer S25型漩涡振荡仪 德国Ika公司;IS-RSDS型台式全温恒温振荡器 美国Crystal公司;CTHI-100B型恒温恒湿箱 美国Stik公司;DRP-9082型电热恒温培养箱上海森信实验仪器有限公司;JY-SPFT型电泳槽、JY600型电泳仪 北京君意东方电泳设备有限公司;BXM-30R型立式压力蒸汽灭菌器 上海博讯实业有限公司;VS-1300L-U型超净工作台 苏州安泰空气技术有限公司。
1.3 方法
1.3.1 引物设计
根据文献[12-20]中提到的3种菌的特异性靶基因,即沙门氏菌的invA基因、单增李斯特菌的PrfA基因和金黄色葡萄球菌的nuc基因,应用Primer premier 5.0设计引物,并对产物长度、T
m值、G-C含量及退火温度等进行优化,最终得到3对特异性引物(表1)。3 对引物由南京金斯瑞生物科技有限公司合成,扩增片段大小依次为284、388、484 bp。
表 1 多重PCR引物序列
Table 1 Primers used for multiplex PCR
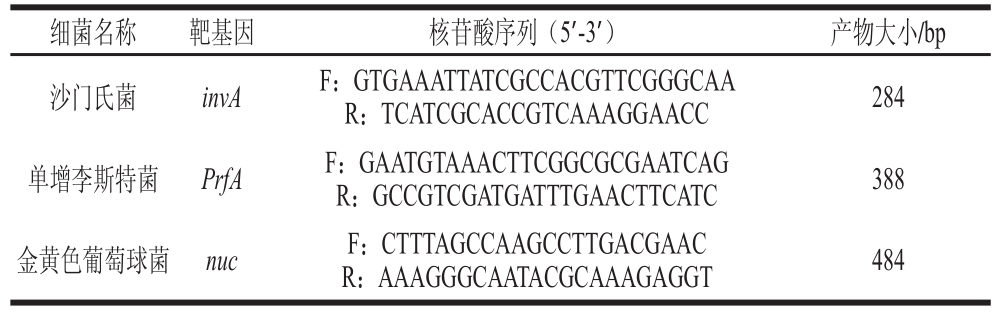
细菌名称靶基因核苷酸序列(5′-3′)产物大小/bp沙门氏菌invAF:GTGAAATTATCGCCACGTTCGGGCAA R:TCATCGCACCGTCAAAGGAACC284单增李斯特菌PrfAF:GAATGTAAACTTCGGCGCGAATCAG R:GCCGTCGATGATTTGAACTTCATC388金黄色葡萄球菌nucF:CTTTAGCCAAGCCTTGACGAAC R:AAAGGGCAATACGCAAAGAGGT484
1.3.2 细菌的培养和DNA模板的制备
将3种标准菌株在TSA-YE培养基上划线,培养复壮18 h,再转种于BHI培养基中,随后取100 μL添加至食品样中,接种于NB培养基中,37 ℃静置过夜培养。取菌液1 mL置于1.5 mL离心管中,12 000 r/min离心3 min后,弃上清,收集菌体,然后依据细菌基因组DNA提取试剂盒的步骤提取DNA。
1.3.3 单重PCR反应体系的建立
单重PCR扩增采用25 μL反应体系:10×PCR缓冲液2.5 μL、2.5 mmol/L dNTPs 2.0 μL、2.5 U/μL的DNA聚合酶0.5 μL、上、下游引物各0.5 μL、模板1 μL,加ddH
2O补足至25 μL。反应条件为:94 ℃/5 min;94 ℃/45 s;64 ℃/30 s;72 ℃/30 s;共计35 个循环;72 ℃/10 min;4 ℃保存。扩增产物采用2.5 g/100 g琼脂糖、100 V电压进行电泳检测,割胶回收后的产物送南京金斯瑞生物科技有限公司测序鉴定。
1.3.4 多重PCR的反应条件的优化
以沙门氏菌、单增李斯特菌和金黄色葡萄球菌提取的DNA为模板,与各种反应试剂混合在一起进行多重PCR反应条件和体系的优化,以建立最佳的m-PCR方法。优化的条件主要包括引物浓度配比、退火温度、退火时间、DNA聚合酶量等。
1.3.5 灵敏度检测
沙门氏菌、单增李斯特菌和金黄色葡萄球菌的标准菌株培养增菌后接种于烟熏火腿,过夜培养后计数,将3种已测浓度的菌液等体积混合并稀释成9 个梯度(10
8~10
0),稀释液2 mL提取DNA,单重PCR以各自提取DNA为模板进行检测,多重PCR以同梯度等体积的DNA为模板进行检测。
1.3.6 实际样品的检测
自江苏雨润肉食品有限公司采集生产50 d左右于4 ℃贮藏的低温肉制品2 种8 份(表2)。在生物安全柜内剪取20 g样品稀释于80 mL含0.1 g/100 g的蛋白胨-0.85 g/100 g的生理盐水中,摇床振荡30 min,4℃条件下4 000 r/min离心10 min,取上清液20 mL于4 ℃条件下10 000 r/min离心20 min,取沉淀置于1.5 mL的离心管,然后依据细菌基因组DNA提取试剂盒的步骤提取DNA。
表 2 检测的样品
Table 2 Samples tested in this study

产品名称牛肉火腿牛肉火腿牛肉火腿烟熏火腿烟熏火腿烟熏火腿烟熏火腿烟熏火腿贮藏时间/d4951525346495051
2 结果与分析
2.1 单重PCR特异性实验
3种目标菌进行单重PCR扩增,验证引物与模板之间的特异性,实验结果如图1所示。
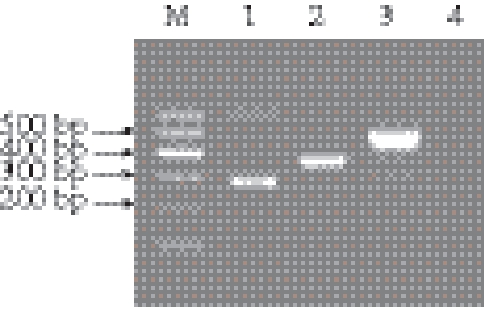
图 1 单重PCR产物的凝胶电泳
Fig. 1 Agarose gel electrophoresis of simplex PCR products
M. DNA Marker;1. Salmonella spp.;2. L.monocytogenes;3.Staphylococcus aureus;4.阴性对照。
由图1可知,泳道1的扩增条带位于200~300 bp之间,与沙门氏菌PCR扩增产物片段长度的实验预期相符;泳道2的扩增条带位于300~400 bp之间,与单增李斯特菌PCR扩增产物片段长度的实验预期相符;泳道3的扩增条带位于400~500 bp之间,与金黄色葡萄球菌PCR扩增产物片段长度的实验预期相符;引物之间无交叉反应。将PCR扩增后的3个目的片段测序结果在NCBI中进行BLAST比对分析,比对结果显示,测序结果与NCBI中沙门氏菌相应基因序列的同源性为99%,与单增李斯特菌相应基因序列的同源性为99%,与金黄色葡萄球菌相应基因序列的同源性为99%。
2.2 多重PCR扩增及反应条件的优化
2.2.1 反应温度的优化

图 2 优化退火温度
Fig. 2 Optimization of annealing temperature
M. DNA Marker;0. 阴性对照;1~6. 温度分别为56、58、60、62、64、66 ℃。
不同菌种由于引物、碱基对等的不同造成了退火温度的差异,为确定多重PCR反应最适退火温度,自56 ℃起至66 ℃设置6 个梯度:56、58、60、62、64、66 ℃,电泳结果见图2。在多重PCR扩增中,不同的温度下,单增李斯特菌和金黄色葡萄球菌均有明显的条带,但沙门氏菌的目的条带亮度较弱且模糊不清,不同的退火温度下,都没有呈现出较好的实验结果,接下来对引物浓度进行优化。
2.2.2 引物浓度的优化
图2中沙门氏菌的条带较为模糊,故对沙门氏菌的PCR扩增条件进行优化,调整引物用量,将沙门氏菌的上、下游引物增加为1.0 μL或者减少为0.25 μL,单增李斯特菌和金黄色葡萄球菌的引物量及其他均不变,ddH
2O补足至25 μL,退火温度选择沙门氏菌单独PCR扩增较好的64 ℃,电泳结果见图3。
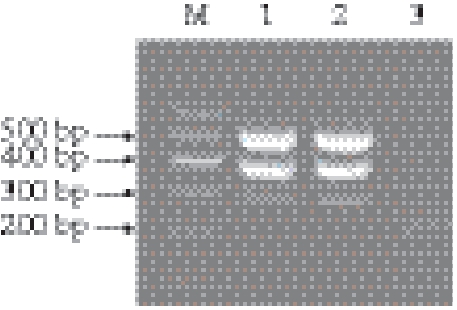
图 3 引物浓度的优化
Fig. 3 Optimization of primer concentration
M. DNA Marker;1~2. 引物用量分别为1.0、0.25 μL;3. 阴性对照。
由图3可知,增加或减少沙门氏菌的引物量时,结果并未得到明显改善,电泳条带亮度依然较弱,接下来对DNA模板做进一步优化。
2.2.3 模板浓度的优化
图3中沙门氏菌的条带模糊不清,推测为DNA模板量不足引起,故增加沙门氏菌DNA模板为2.0 μL和3.0 μL,其余试剂和条件的量均不变,ddH
2O补足至25 μL,退火温度选择为64 ℃。PCR产物电泳结果见图4。
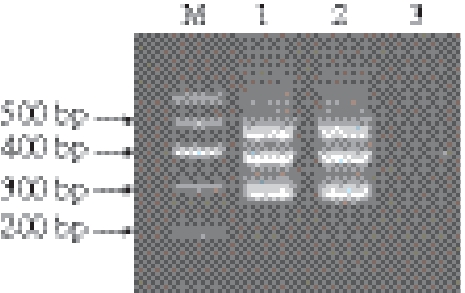
图 4 模板浓度的优化
Fig. 4 Optimization of template concentration
M. DNA Marker;1~2. DNA模板量分别为2.0、3.0 μL;3. 阴性对照。
由图4可知,3种菌的目的条带清晰可见,在DNA模板为2.0 μL时,多重PCR检测肉制品中沙门氏菌、单增李斯特菌和金黄色葡萄球菌具有较好的效果,此时,沙门氏菌的灵敏度稍低于单增李斯特菌和金黄色葡萄球菌。
2.2.4 多重PCR体系灵敏度
表 3 多重PCR灵敏度
Table 3 Sensitivity of multiplex PCR
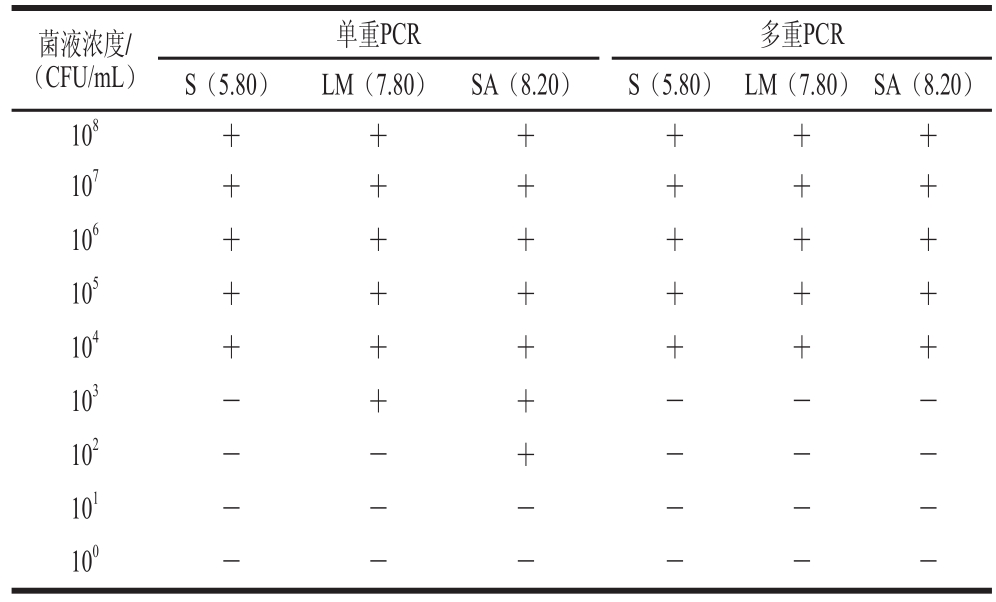
注:S. 沙门氏菌;LM. 单增李斯特菌;SA. 金黄色葡萄球菌;+. 有PCR条带;-. 无PCR条带。
菌液浓度/(CFU/mL)单重PCR多重PCR S(5.80)LM(7.80)SA(8.20)S(5.80)LM(7.80)SA(8.20)10
8++++++10
7++++++10
6++++++10
5++++++10
4++++++10
3-++---10
2--+---10
1------10
0------
由表3可知,多重PCR灵敏度结果显示,单重PCR中3种目的菌的最低检出限为金黄色葡萄球菌8.20×10
2CFU/mL,多重PCR的最低检测限为10
4CFU/mL显著低于单重PCR,在多重PCR反应体系中,随着引物、模板种类的增多和量的增加,结合率反而呈下降趋势
[14]。
对比单重和多重PCR的检出限可知后者灵敏度显著下降,对多重PCR反应体系的引物浓度、退火温度、退火时间、DNA聚合酶量等进行优化,最适结果如下:25 μL反应体系:10×PCR缓冲液2.5 μL及2.5 mmol/L dNTPs 2.0 μL、2.5 U/μLDNA聚合酶0.5 μL、上、下游引物各0.5 μL、单增李斯特菌和金黄色葡萄球菌的模板1.0 μL、沙门氏菌的模板2.0 μL,加ddH
2O补足至25 μL。反应条件为:94 ℃/5 min,94 ℃/45 s,64 ℃/30 s,72 ℃/30 s,扩增35个循环;72 ℃/10 min,4℃保存。扩增产物采用2.5 g/100 g琼脂糖、100 V电压进行琼脂糖凝胶电泳,结果出现3 条清晰的条带,大小分别约为284、388、484 bp。
2.2.5 样品检测
采用优化后多重PCR反应体系和反应条件对贮藏50 d左右的8份样品进行检测,结果如图5所示。

图 5 样品多重PCR检测结果
Fig. 5 PCR detection of real samples
M. DNA Marker;0. 阴性对照;1~5. 牛肉火腿;6~8. 烟熏火腿;9. 阳性对照。
由图5可知,8份样品均未有沙门氏菌、单增李斯特菌和金黄色葡萄球菌的检出。
3 讨 论
分子生物学方法因其简单、快速、灵敏度高等优点,在食源性致病菌检测中已得到广泛的应用,最常见的即是PCR技术,常规PCR一次检测一种病原菌,但样品中往往存在多种致病菌,多重PCR(m-PCR)一次可检测2 种或2 种以上致病菌
[15],在实际推广应用中更加方便有效。
多重PCR并非把普通PCR反应体系进行简单的混合,反应体系会受到诸多因素的影响。影响第一因素就是靶基因的选择,沙门氏菌选择侵袭因子invA为靶基因,前人研究
[16-17]表明该基因在属内均存在、属间特异性表达;单增李斯特菌选择其重要的转录调节因子PrfA为靶基因,该基因对毒力有重要的影响
[18-19];金黄色葡萄球菌选取特异性基因耐热核酸酶nuc基因
[20],此3 种靶基因序列均高度保守,增加扩增结果的特异性和准确性。影响的第二因素是引物设计,前人研究
[21]表明稍长的引物和稍高的解链温度,均可增加m-PCR扩增产物的特异性,本研究退火温度优化为较高的64℃与此实验结果一致。除此外多重PCR反应体系还受到其他多种因素的影响,例如dNTP、Mg
2+、Taq酶以及引物间和模板间的相互影响等
[22]。鉴于此,优化最适反应体系,具体如下:总体积为25 μL,10×PCR缓冲液2.5 μL、2.5 mmol/L d NTPs 2.0 μL、2.5 U/μLDNA聚合酶0.5 μL、上、下游引物各0.5 μL、单增李斯特菌和金黄色葡萄球菌的模板1.0 μL、沙门氏菌的模板2.0 μL。退火温度、模板浓度等条件优化有利于m-PCR检测目标菌株,以便得到更加明确、清晰的条带和更为精确的检测限。m-PCR方法具有灵敏度高的优点,舒畅等
[23]利用m-PCR检测3 种致病菌的灵敏度为10
4CFU/mL,王虎虎等
[13]研究三重PCR同时检出的鸡肉中3 种致病菌最低检测限为10
3CFU/mL。本研究m-PCR检测的最低限为10
4CFU/mL相对偏低,推测为两方面原因,一与基因组DNA纯度和浓度有关,Stefanova等
[24]比较十六烷基三甲基溴化铵(hexadecyltrimethylammonium bromide,CTAB)法和3 种试剂盒法提取DNA,发现试剂盒方法提取的DNA浓度和纯度均偏低,从而导致检测灵敏度略低;二与烟熏火腿中成分介质过多有关,某些成分可抑制PCR反应,Wilson
[25]研究表明肉制品中的脂肪可导致PCR灵敏度降低。本实验下一步将采取CTAB等方法提取基因组DNA并对比优化以提高检测灵敏度;检测脂肪、盐、蛋白和配料等肉制品中介质对PCR灵敏度的影响。但由于多数致病菌的感染剂量均大于10
3CFU/mL
[26],故本结果对实际样品检测具有重要的参考意义。多重PCR中目的片段在扩增反应时相互拮抗导致检测的灵敏度低于单重PCR,利用优化后的m-PCR体系对贮藏50 d左右的8 份样品进行多重PCR检测,均未检测出阳性结果,与实际情况相符。
本研究通过优化多重PCR反应条件,建立了低温肉制品中3种食源性致病菌多重PCR快速检测方法,检测结果有较强的特异性、较高的灵敏度、操作省时省力、成本低,符合当前食品快速检测的需要,具有较强的实用价值,在当前的食品安全检测工作中具有较强的推广和应用前景。
参考文献:
[1] 徐方旭, 刘诗扬, 兰桃芳, 等. 食源性致病菌污染状况及其应对策略[J]. 食品研究与开发, 2014, 35(1): 98-101. DOI:10.3969/ j.issn.1005-6521.2014.01.027.
[2] 徐君飞, 张居作. 2001—2010年中国食源性疾病暴发情况分析[J].中国农学通报, 2012, 28(27): 313-316.
[3] LI M Y, ZHOU G H, XU X L, et al. Changes of bacterial diversity and main fl ora in chilled pork during storage using PCR-DGGE[J]. Food Microbiology, 2006, 23(7): 607-611. DOI:10.1016/j.fm.2006.01.004.
[4] ABUBAKAR I, IRVINE L, ALDUS C F, et al. A systematic review of the clinical, public health and cost-effectiveness of rapid diagnostic tests for the detection and identification of bacterial intestinal pathogens in faeces and food[J]. Health Technology Assessment, 2007, 11(36): 1-216.
[5] DALVIT C, MARCHI M, CASSANDRO M, et al. Genetic traceability of livestock products: a review[J]. Meat Science, 2007, 77(4): 437-449.
[6] SEVERGNINI M, CREMONESII P, CONSOLANDI C, et al. Advances in DNA microarray technology for the detection of foodborne pathogens[J]. Food and Bioprocess Technology, 2011, 4(6): 936-953. DOI:10.1007/s11947-010-0430-5.
[7] EDWARDS M C, GIBBS R A. Multiplex PCR: advantages, development, and applications[J]. Genome Research, 1994, 3(4): 65-75.
[8] ROY A, FAYAD A, BARTHE G, et al. A multiplex polymerase chain reaction method for reliable, sensitive andaimultaneousdetection of multiple viruses in citrus trees[J]. Journal of Virological Methods, 2005, 129(1): 47-55.
[9] BABU L, REDDY P, MURALI H S, et al. Optimization and evaluation of a multiplex PCR for simultaneous detection of prominentfoodborne pathogens of Enterobacteriaceae[J]. Annals of Microbiology, 2013, 63 (4): 1591-1599. DOI:10.1007/s13213-013-0622-0.
[10] SCALLAN E, HOEKESTRA R M, ANGULO F J, et al. Foodborne illness acquired in the united states-major pathogens[J]. Emerging Infectious Diseases, 2011, 17(1): 7-15. DOI:10.3201/eid1701.P11101.
[11] KUMAR A, GROVER S, BATISH V K. Exploring specific primers targeted against different genes for a multiplex PCR fordetection of Listeria monocytogenes[J]. 3 Biotech, 2015, 5(3): 261-269. DOI:10.1007/s13205-014-0225-x.
[12] 滕要辉, 索标, 艾志录, 等. 速冻食品中沙门氏菌和金黄色葡萄球菌多重PCR检测方法的建立与应用[J]. 食品科学, 2013, 34(8): 140-144. DOI:10.7506/spkx1002-6630-201308028.
[13] 王虎虎, 徐幸莲. 冰鲜鸡肉中致病菌三重PCR检测方法的建立[J].中国农业科学, 2010, 43(17): 3608-3615.
[14] 王慧, 朱瑞良, 谭燕玲, 等. 多重PCR检测三种重要食源性致病菌方法的建立及应用[J]. 中国农业科学, 2011, 44 (11): 2334-2340. DOI:10.3864/j.issn.0578-1752.2011.11.016.
[15] CATTOIR V, POIREL L, ROTIMI V, et al. Multiplex PCR for detection of plasmid-mediated quinolone resistance qnrgenes in ESBL-producing enterobacterialisolates[J]. Journal of Antimicrobial Chemotherapy, 2007, 60(2): 394-397. DOI:10.1093/jac/dkm204.
[16] RAHN K, GRANDIS S A D, CLARKE R C, et al. Amplifi cation of an invA genesequence of Salmonella typhimurium by polymerase chain reaction asa specific method of detection of Salmonella[J]. Molecular and Cellular Probes, 1992, 6: 271-279. DOI:10.1016/0890-8508(92)900002-F.
[17] DAVID R L, MARTA H, TERESA E, et al. A rapid and directreal time PCR-based method for identifi cation of Salmonella spp[J]. Journal of Microbiological Methods, 2003, 54: 381-390.
[18] MAUDER N, ECKE R, MERTINS S, et al. Sapies- specifi c differences in the activity of PrfA, the key regulator of listerial virulence genes[J]. Journal of Bacteriology, 2006, 188(22): 7941-7956.
[19] SORTTI M, MONZ H J, LACHAREM L, et al. The PrfA virulence regulon[J]. Microbes and Infection, 2007, 9(10): 1196-1207.
[20] BRASHER C W, DEPAOLA A, JONES D D, et al. Detection ofmicrobial pathogens in shellfish with multiplex PCR[J]. Current Microbiology, 1998, 37: 101-107. DOI:10.1007/s002849900346.
[21] SETTANNI L, CORSETTI A. The use of multiplex PCR to detect and differentiate food and beverage -associated microorganisms: a review[J]. Journal Microbiology Methods, 2007, 69(1): 1-22.
[22] MARKOULATOS P, SIAFAKAS N, MONCANY M. Multiplex polymerase chain reaction: a practical approach[J]. Journal of Clinical Laboratory Analysis, 2002, 16(1): 47-51. DOI:10.1002/jcla.2058.
[23] 舒畅, 姜琛璐, 钟慈, 等. 三种食源性致病菌多重PCR检测方法的建立[J]. 食品工业科技, 2014, 35(12): 49-54.
[24] STEFANOVA P, TASEVA M, GEORGIEVAT, et al. Amodified CTAB method for DNA extraction from soybean and meat products[J]. Biotechnology and Biotechnological Equipment, 2013, 27(3): 3803-3810.
[25] WILSON I G. Inhibition and facilitation of nucleic acid amplification[J]. Applied and Environment Microbiology, 1997, 63(10): 3741-3751.
[26] KONG R Y C, LEE S K Y, LAW T W F, et al. Rapiddetected of six types of bacterial pathogens in marine waters by multiplex PCR[J]. Water Research, 2002, 36(11): 2802-2812.
Multiplex Polymerase Chain Reaction Assay for Rapid Detection of Three Foodborne Pathogens in Low-Temperature Meat Products
LI Xinfu
1,2, WANG Zhouping
1, LI Cong
1,2, FANG Lingli
1, ZHAO Ying
2, XU Baocai
1,2,3,*
(1.School of Food Science and Technology, Jiangnan University, Wuxi 214122, China; 2.The State Key Laboratory of Meat
Processing and Quality Control, Jiangsu Yurun Meat and Food Co. Ltd., Nanjing 211806, China; 3.Jiangsu Collaborative Innovation Center of Meat Productionand Processing, Quality and Safety Control, Nanjing 210095, China)
Abstract:Salmonella spp., Listeria monocytogenes (L. monocytogenes) and Staphylococcus aureus (S. aureus) are 3 species of bacteria which can easily contaminate low-temperature meat products. The objective of this study was to develop a rapid and effi cient method for the simultaneous detection of the bacterial pathogens using multiplex polymerase chain reaction (m-PCR). The specifi c primers for the invA gene of Salmonella spp., the PrfA gene of L. monocytogenes and the nuc gene of S. aureus were designed and their specifi city was determined. After enrichment culture, the 3 strains were inoculated to lowtemperature meat products and incubated overnight for evaluating the sensitivity of the detection method. Optimization of the PCR system and its application to the detection of real samples were carried out. Our experimental results showed that the optimum PCR conditions were determined as follows: using a reaction system containing 2.5 μL of 10 × PCR buffer, 2.0 μL of 2.5 mmol/L dNTPs, 0.5 μL of 2.5 U/μL DNA polymerase, 0.5 μL of upstream primer, 0.5 μL of downstream primer, 1.0 μL of L. monocytogenes template, 1.0 μL of S. aureus template or 2.0 μL of Salmonella spp. template brought with ddH
2O up to 25 μL, and an annealing temperature of 64 ℃. The multiplex PCR method was applicable to detect Salmonella spp. in meat products with the advantages of rapidity, high sensitivity and specifi city and simplicity.
Key words:low-temperature meat products; multiplex polymerase chain reaction (m-PCR); rapid detection; foodborne pathogens
DOI:10.7506/rlyj1001-8123-201702009
中图分类号:TS251.7
文献标志码:A
文章编号:1001-8123(2017)02-0045-06
收稿日期:2016-08-06
基金项目:国家自然科学基金面上项目(31571909);“十三五”国家重点研发计划重点项目(2016YFD0400703-05)
作者简介:李新福(1982—),男,博士研究生,研究方向为肉品质量安全控制。E-mail:lixinfu316@126.com
*通信作者:徐宝才(1973—),男,教授级高级工程师,博士,研究方向为肉品质量安全控制。E-mail:baocaixu@163.com
引文格式:
李新福, 王周平, 李聪, 等. 低温肉制品中3 种食源性致病菌多重聚合酶链式反应快速检测方法的建立[J]. 肉类研究, 2017, 31(2): 45-50. DOI:10.7506/rlyj1001-8123-201702009. http://www.rlyj.pub
LI Xinfu, WANG Zhouping, LI Cong, et al. Multiplex polymerase chain reaction assay for rapid detection of three foodborne pathogens in low-temperature meat products[J]. Meat Research, 2017, 31(2): 45-50.
DOI:10.7506/rlyj1001-8123-201702009. http://www.rlyj.pub